
運動神經元病
概述
運動神經元是指負責將脊髓和大腦發出的信息傳到肌肉和內分泌腺,支配器官活動的神經元。運動神經元病屬於罕見疾病,是一系列以運動神經元改變為突出表現的慢性進行性神經系統變性疾病。目前發病原因尚不明確,病人表現為肌無力、肌萎縮、延髓麻痹等,通常無感覺異常。該病多為中老年發病,病程多為2~6年,大多預後不良,常因呼吸肌受累死於呼吸肌麻痹或肺部感染。
- 就診科別:
- 神經內科
- 英文名稱:
- moto rneuron disease,MND
- 疾病別稱:
- 肌萎縮側索硬化
- 是否常見:
- 否
- 是否遺傳:
- 部分遺傳
- 併發疾病:
- 肺部感染、營養不良、褥瘡
- 治療周期:
- 終身間歇性治療
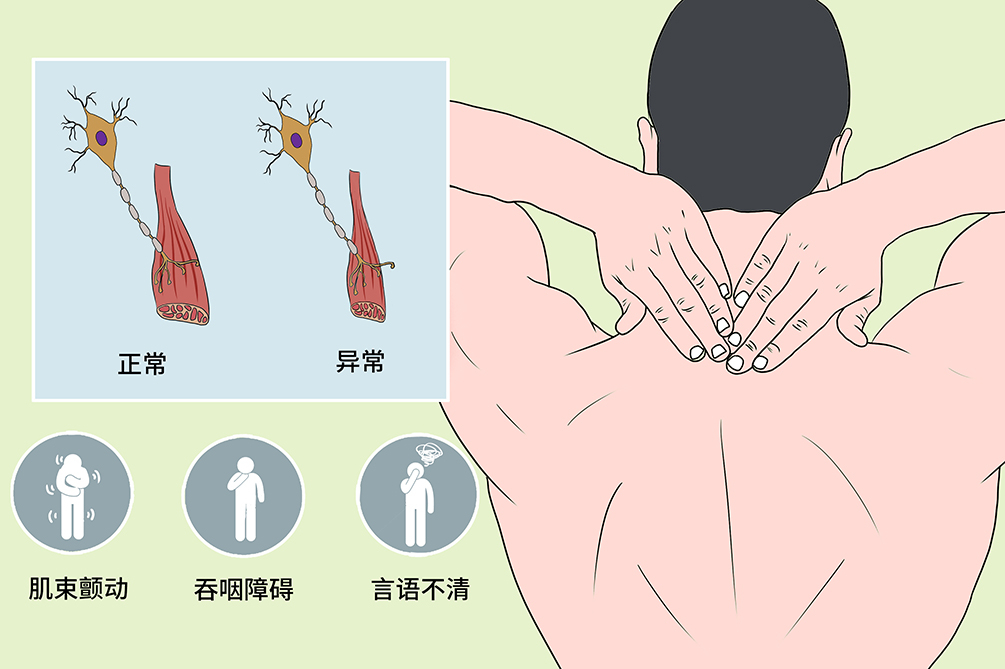
- 臨床症狀:
- 肌無力、肌萎縮、肌束顫動、吞咽障礙、言語不清
- 好發人群:
- 中老年男性、外傷史者、過度體力勞動者
- 常用藥物:
- 利魯唑
- 常用檢查:
- 肌電圖、腦脊髓液檢查、血液檢查、CT、磁振造影檢查、肌肉切片檢查
下運動神經元型
多於30歲左右發病。通常以手部小肌肉無力和肌肉逐漸萎縮起病,可波及一側或雙側,或從一側開始以後再波及對側。顱神經損害常以舌肌最早受侵,出現舌肌萎縮,伴有顫動,以後齶、咽、喉肌,咀嚼肌等亦逐漸萎縮無力,以致病人構音不清,吞咽困難,咀嚼無力等。包括進行性脊肌萎縮和進行性延髓萎縮。
上運動神經元型
表現為肢體無力、發緊、動作不靈。因病變常先侵及下胸髓的皮質脊髓束,故症狀先從雙下肢開始,以後波及雙上肢,且以下肢為重。本症稱原發性側索硬化症,臨床上較少見,多在成年後起病,一般進展甚為緩慢。
上、下運動神經元混合型
通常以手肌無力、萎縮為首發症狀,一般從一側開始以後再波及對側,隨病程發展出現上、下運動神經元混合損害症狀,稱為肌萎縮側索硬化症。病程末期,全身肌肉消瘦萎縮,以致抬頭不能,呼吸困難,臥床不起。
病因
運動神經元病確切的病因和發病機制迄今未明。可能為各種原因引起神經系統有毒物質堆積,特別是自由基和興奮性胺基酸的增加,損傷神經細胞而致病。
遺傳因素
運動神經元病大多為散發,少數有家族史,遺傳方式主要為常染色體顯性遺傳。最常見的致病基因是21號染色體上的銅(鋅)超氧化物歧化酶(SOD-1)基因。
感染和免疫
有研究人員認為該病的發病可能與脊髓灰質炎病毒、腸病毒、人類免疫缺陷病毒(HIV)有關,當感染侵及神經元,病人會出現相應的臨床表現。
金屬元素
運動神經元病發病可能與某些金屬,如鉛、汞、鋁等有關,環境中金屬元素含量的差異也可能是某些地區地理性高發病率的原因。
營養障礙
有研究人員發現肌萎縮側索硬化(ALS)的病人血漿中維他命B1及單磷酸維他命B1均有減少,這些因素可能會導致神經元的正常代謝障礙,進而導致肌萎縮側索硬化。
神經遞質
ALS病人的腦脊髓液中抑制性神經遞質GABA水平明顯降低,而去甲腎上腺素升高,病情越嚴重這種變化越明顯,也有研究表明興奮性胺基酸(主要是穀氨酸和天門冬氨酸)的神經細胞毒性作用在ALS發病中起著重要作用。
外傷
人體在外傷後可能會導致神經的損傷,進而出現相應的症狀,且運動神經元病的病人多曾有過不同程度的外傷史。
過度體力勞動
有資料顯示礦工、重體力勞動者等患運動神經元病的機率更大,因為通過過度的體力勞動,機體內的很多代謝產物不能及時排除體外,同時神經的功能代謝也會受到不同程度的損害。
運動神經元病是一種罕見疾病,年發病率為(1.5~2.7)/10萬,患病率為(2.7~7.4)/10萬。多在中年發病,男性多於女性,男女患病比例為(1.2~2.5):1,約5%~10%的運動神經元病病人有家族史。
中老年男性
該病多為中老年發病,男性多於女性。
有外傷史者
據統計運動神經元病病人大多都有不同程度的外傷史。
過度體力勞動者
重體力勞動者患運動神經元病的機率會增加。
症狀
運動神經元病通常起病隱匿,病程進展緩慢。由於損傷部位不同,臨床表現為肌無力、肌萎縮、延髓麻痹等症狀的不同組合。不少病例先出現一種類型的表現,隨後又出現另一類型的表現,最後演變成ALS。因此在疾病初期有時比較難確定屬於哪種類型。
肌萎縮側索硬化
常見的首發症狀為一側或雙側手指活動笨拙、無力,隨後出現手指小肌肉萎縮,逐漸發展至前臂、上臂及肩部肌群。隨著病程的延長,肌無力和萎縮擴展至軀幹和頸部,最後累及面肌和咽喉肌。少數病例肌萎縮和無力從下肢或軀幹開始。受累部位常有明顯肌束顫動。
進行性肌萎縮
常見首發症狀為單手或雙手小肌肉萎縮、無力,逐漸波及前臂、上臂及肩部肌群。受累肌肉萎縮明顯、肌肉張力降低,可見肌肉顫動。
進行性延髓麻痹
主要表現為進行性發音不清、吞咽困難、飲水嗆咳、咀嚼無力。舌肌明顯萎縮,並有肌束顫動,唇肌、咽喉肌萎縮。本類型病情進展較快,多在1~2年內因呼吸肌麻痹或肺部感染而死亡。
原發性側索硬化
常見首發症狀為雙下肢對稱性僵硬、乏力,行走呈剪刀步態。緩慢進展,逐漸累及雙上肢。一般無肌萎縮和肌束顫動、無感覺障礙。該類型進展慢,可存活較長時間。
有一小部分運動神經元病的病人出現運動系統以外的表現,如痴呆、感覺異常、大小便功能障礙等,也有少部分病人出現眼外肌運動障礙。
肺部感染
運動神經元病末期因呼吸肌受累極易出現呼吸不順、咳嗽反射減弱,分泌物不易排出,從而增加肺部感染的風險,甚至進一步造成呼吸衰竭;另外延髓麻痹會導致飲水嗆咳的出現,增加吸入性肺炎的發生機率。
營養不良
當病變累及延髓時會出現吞咽功能障礙,影響正常飲水、進食,從而導致營養不良、脫水的出現。
褥瘡
當病人病程進展,肌無力發展至影響自主活動時,如果缺少必要的翻身、擦洗等護理工作時,極易導致褥瘡的出現。
看醫
儘管運動神經元病仍是一種無法治癒的疾病,但有許多方法可以改善病人的生活質量,應初期診斷、初期治療,儘可能加強護理、預防各種併發症、延長生存期。
- 如果逐漸出現肢體無力、肌肉萎縮、吞咽困難、發音不清等症狀,尤其是有運動神經元病家族史的人群,需要儘快行肌電圖檢查排查運動神經元病的可能。
- 如果是已經確診運動神經元病的病人,若出現營養障礙、呼吸不順、感染等症狀時,應立即就醫。
- 大多病人優先考慮去神經內科就診。
- 若病人出現其他嚴重不適反應或併發症,如肺部感染等,可到相應科別就診,如呼吸內科等。
- 目前都有什麼症狀?(如肢體乏力、活動笨拙、肌肉萎縮等)
- 症狀是突發的還是緩慢進展的?
- 這種症狀是什麼時候出現的?
- 是否有家族史?
- 既往有無其他的病史?
肌電圖
肌電圖和神經傳導速度測定
是診斷運動神經元病最有價值的手段,特別是在疾病初期,尤其具有診斷和鑑別診斷的價值。
運動誘發電位(錐體束檢查)
此項檢查為運動神經元病提供上運動神經元受累的客觀依據。
單纖維肌電圖檢查
通過特殊的針電極來記錄非常小的單個肌纖維的動作單位。
腦脊髓液檢查
腦脊髓液壓力檢查結果常為正常或偏低,腦脊髓液檢查正常或蛋白有輕度增高,免疫球蛋白可能增高。
血液檢查
血液常規檢查結果為正常。血清肌酸磷化酶活性正常或者輕度升高而其同工酶不高。免疫功能檢查可能出現異常。
CT和磁振造影檢查
主要用於鑑別診斷排除其他結構性病變導致的錐體束或下運動神經元損害。
肌肉切片檢查
可見神經源性肌萎縮的病理改變,並非診斷MND的常規檢查項目,僅在臨床表現不典型或診斷困難,需要與其他疾病鑑別時選擇。
根據中年以後隱匿起病,慢性進行性加重的病症,臨床主要表現為上、下運動神經元損害所致的肌無力、肌萎縮、延髓麻痹等,無感覺障礙,肌電圖呈神經源性損害,腦脊髓液正常,影像學無明顯異常,即可做出臨床診斷。
頸椎病
頸椎病可有手部肌肉萎縮,壓迫脊髓時還可表現出上、下運動神經元病變的症狀和體徵,亦可呈慢性進行性病程,與運動神經元病難以鑑別。但是頸椎病常伴有上肢或肩部疼痛,也常有根性分布的感覺障礙,無延髓麻痹的表現。頸椎X線片、CT或磁振造影提示頸椎骨質增生、椎間孔變窄、椎間盤變性或脫出,甚至脊膜囊受壓,有助於鑑別。
延髓和脊髓空洞症
臨床上也常有雙手小肌肉萎縮、肌束顫動。但該病有典型的節段性分離性感覺障礙,磁振造影可顯示延髓或脊髓空洞,有助於鑑別。
良性肌束顫動
高達70%的正常人有時可出現不同程度的自發性粗大的肌束顫動,但無肌無力和肌萎縮,肌電圖檢查也正常,兩者可以根據肌電圖覺得檢查結果進行鑑別。
治療
目前仍缺乏能夠有效逆轉或控制病情發展的藥物。運動神經元病致病因素多樣且互相影響,故其治療必須是多種方法的聯合應用。運動神經元病的治療包括病因治療、對症治療和各種非藥物治療。
- 當病人出現肌肉痛性痙攣,可對症採用解痙、止痛藥物治療。
- 當病人病情進展,出現呼吸衰竭時,可儘早採用無創呼吸機輔助呼吸,或根據具體情況選擇是否行氣管切開並機械通氣。
利魯唑
抑制穀氨酸釋放的作用,有可能延緩病程、延長延髓麻痹病人的生存期,但對病人的肌力和生活質量沒有顯著改善。
運動神經元病的病人很有可能存在焦慮、憂鬱的情緒,應加強心理疏導,必要時可給予抗焦慮、憂鬱藥物。如病人存在睡眠障礙應予心理治療,並視情況給予樂比克、 Zolpidem 等助眠藥物。
- 近來有研究表明,神經幹細胞移植、基因治療等方法可能對運動神經元病有一定效果,但目前仍處於研究階段,臨床應用並不廣泛。
- 可以應用針灸、按摩、理療等中醫療法改善病人肢體狀況。
預後
運動神經元病的預後因不同的疾病類型和發病年齡而有所不同。早診斷、早治療能夠延長病人生存期,改善生活質量。
運動神經元病目前尚不能治癒。
- 原發性側索硬化進展緩慢、預後良好,可存活較長時間。
- 部分進行性肌萎縮病人的病情可以維持較長時間穩定,但不會改善。
- 肌萎縮側索硬化、進行性延髓麻痹以及部分進行性肌萎縮病人的預後差,病情持續性進展,多於5年內死於呼吸肌麻痹或肺部感染。
運動神經元病剛開始發病時,至少每3~6個月複查一次;隨著病情進展,如出現吞咽障礙、肺部感染等症狀時可能會出現頻繁住院,甚至長期住院治療。
飲食
目前對於運動神經元病並無針對性飲食宜忌,均衡飲食即可。
照護
運動神經元病病人的護理應時刻注意觀察病人的營養狀態、吞咽功能和呼吸狀態等,關心病人的心理狀況,保持環境乾淨整潔,減少併發症的出現。
- 按照醫囑用藥,了解藥物可能發生的不良反應,服藥期間定期複查血液常規及肝腎功能。
- 根據自身的病情,病人在醫生指導下進行一些適當的運動。
- 對於長期臥床的病人應保持周圍環境的乾淨、整潔,家屬或護理人員應每天按時幫助病人翻身、拍背、按摩、擦洗,減少感染、褥瘡等併發症的出現。
- 對存在吞咽困難的病人應放緩進食速度,儘量避免誤吸的出現。
時刻注意觀察病人的營養狀態、吞咽功能和呼吸狀態等。
保持良好心態,家屬應時刻關注病人的情緒變化,給予病人合適的心理支持,適時對病人進行心理疏導。
儘量避免接觸重金屬、化學物質、有毒物等。
預防
由於運動神經元病的發病原因不明,因此目前還沒有特異而有效的預防方法。但保持良好的生活習慣對預防疾病發生有益處。
- 避免將自己暴露於鉛、汞、鋁等金屬環境中,儘量避免過度重體力勞動。
- 儘量避免外傷的出現。