
先天性腎上腺增生症
概述
先天性腎上腺增生症是一組常染色體隱性遺傳病,由於腎上腺皮質類固醇激素合成過程中某種酶的先天缺陷,引起腎上腺皮質激素合成不足,經負反饋作用促使下丘腦、垂體分泌促腎上腺皮質激素釋放激素和促腎上腺皮質激素增加,導致腎上腺增生症和代謝失調。臨床主要表現為不同程度的腎上腺皮質功能減退、性腺發育異常、伴或不伴水鹽代謝失調與高血壓,以糖和鹽皮質激素替代治療為主,療效和預後取決於酶缺陷的程度、就診的早晚及開始治療的早晚 病人的依從性,能否堅持服藥和定期隨診等。
- 就診科別:
- 內分泌科、泌尿外科
- 英文名稱:
- congenital adrenal hyperplasia,CAH
- 疾病別稱:
- 原發性腎上腺增生症、先天性柯興氏症候群
- 是否常見:
- 否
- 是否遺傳:
- 是
- 併發疾病:
- 腎上腺危象、甲狀腺機能低下、心肌損害
- 治療周期:
- 長期持續性治療
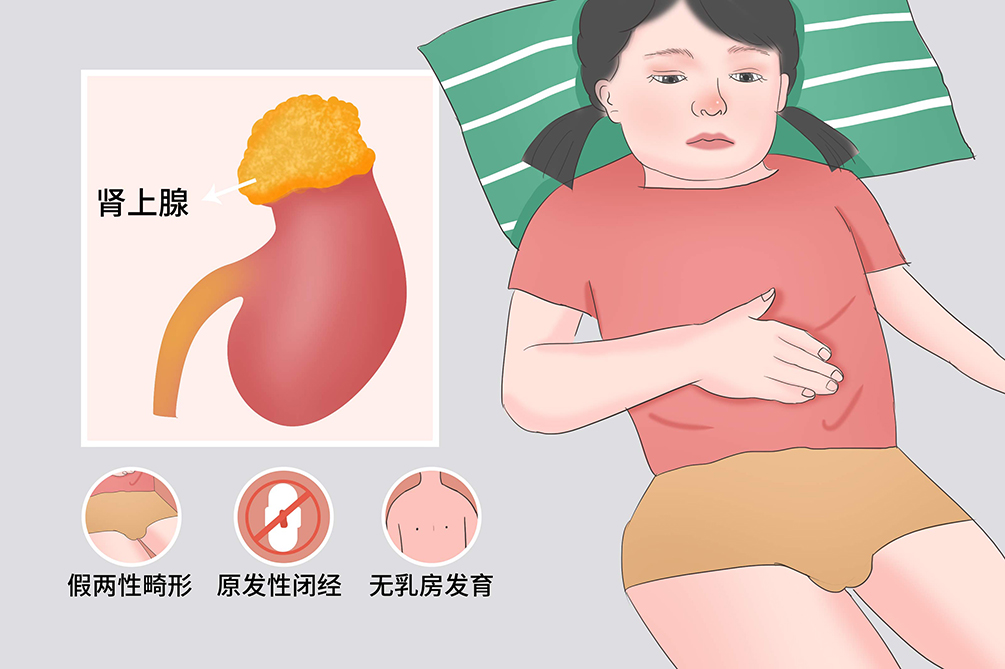
- 臨床症狀:
- 假兩性畸形、大陰唇融合、陰蒂肥大、外生殖器明顯增大、陰囊增大
- 好發人群:
- 有家族遺傳史者
- 常用藥物:
- 氫化可的松、醋酸氫化可的松、9a-氟氫可的松
- 常用檢查:
- 尿 17- 羥類固醇、去氧皮質酮及睪固酮測定、血皮質醇、血電解質檢查、染色體檢查
病因
先天性腎上腺增生症的病因主要包括21-羥化酶缺乏症、11β-羥化酶缺乏症、3β-羥脫氫酶缺乏症、17α-羥化酶缺乏症、膽固醇碳裂解酶缺乏症、類脂性腎上腺增生症等類型。其中21-羥化酶缺乏症最常見,約占總數的 90%~95% , 11β-羥化酶缺乏症次之,約占 7%,再其次為3β-羥脫氫酶缺乏症,17α-羥化酶缺乏症和膽固醇碳裂解酶缺乏症則十分罕見。
21-羥化酶缺乏
為最常見的一種類型,約占先天性腎上腺增生症病人的90%~95%。該酶的缺陷使最終產物皮質醇和鹽皮質激素產生不足和17羥孕酮的堆積,伴雄激素分泌過多。
11β-羥化酶缺乏
為第二種常見類型,約占5%~8%。由於CYP11β基因突變,腎上腺不能進一步合成皮質醇,11-去氧皮質醇增多和11-去氧皮質酮、脫氫異雄酮和4-雄烯二酮分泌增多,引起女性男性化和男性假性性早熟。
17α-羥化酶缺乏
是較少見的類型 、占本病的1%。CYP17缺陷使腎上腺、睪丸、卵巢的性激素的合成完全被阻斷。
3β-羥脫氫酶缺乏
是一種危險的類型,皮質醇和鹽皮質激素合成均嚴重受阻。
細胞色素P450氧化還原酶缺乏
是一種罕見的先天性腎上腺增生症,由於細胞色素P450氧化還原酶缺陷,導致類固醇激素合成障礙。
先天性類脂質性腎上腺增生
是最少見和最嚴重的第一型,腎上腺皮質不能合成皮質醇、醛固酮及雄激素等任何一種皮質激素,膽固醇不能被利用,而在細胞內存積。垂體促腎上腺皮質激素促進皮質增生,表現為類脂質細胞的增生。
- 近親婚配:本病為常染色體隱性遺傳,近親婚配使得該病患病率增加。
- 胃腸疾病、感染、圍手術、疼痛、心理壓力、藥物不足、突發事故等易誘發腎上腺危象。
本病是較常見的常染色體隱性遺傳病,發病常有家族性,在同一家族中常表現為同一類型的缺陷。典型的先天性腎上腺增生症發病約為1/10000 ~1/ 20000活產新生兒,同種族先天性腎上腺增生症發病也有很大區別。
本病為較常見的常染色體隱性遺傳病,因此發病常有家族性,在同一家族中常表現為同一類型的缺陷。
症狀
先天性腎上腺增生症以女孩多見,男女之比約為1: 2,其臨床表現取決於酶缺陷的部位及缺陷的嚴重程度。
21-羥化酶缺乏症
典型失鹽型
嘔吐、腹瀉、餵養困難、體重不增、脫水、酸中毒及難以糾正的低血鈉、高血鉀等表現,多在生後1~2週發病。新生兒出生後即可表現為腎上腺危象,如不及時治療,可出現血容量降低、血壓下降和休克。
單純男性化
女性胎兒外生殖器男性化,女性男性化的程度不一,可從單純陰蒂增大常被誤認為男性化不全的男性,到完全性陰囊性尿道下裂伴隱睪的男性。若不及時治療陰蒂可繼續增大,體毛增多,痤瘡出現,女性第二性徵發育不良,甚至無乳房發育和原發性閉經,無生殖能力。男性患兒出生時外生殖器可無明顯異常,易誤診。隨生長過程因高雄激素作用,在嬰幼兒期,陰莖進一步增大,睪丸不大,伴有或不伴有陰毛早現的外周性性早熟,由於性激素過早、長時期的作用,使兒童期生長過速伴骨齡快速增長,骨骺過早閉合致成人矮身材。
不典型的晚髮型
大多出生時無臨床症狀,外生殖器正常,隨年齡增長女性病人可有陰毛早現,月經初潮延遲,月經量過少或部分病人可發生嚴重痤瘡,不明原因的骨骺成熟增速 、生長增快的表現,最後身高矮於父母的平均身高。
11β-羥化酶缺乏症
典型
部分出現高血鈉、低血鉀、鹼中毒和高血容量,又可因皮質醇減少出現皮質功能減低的症狀及雄激素過高的症狀。
不典型
不典型者臨床差異較大,大部分因臉部痤瘡、月經失調來就診,少數有高血壓,大多血壓正常。
17α-羥化酶缺乏症
大部分患兒出現高血壓、高血鈉、低血鉀和鹼中毒,有輕度皮質醇不足的症狀,男性假兩性畸形,男性女性化。女性因雌激素缺乏表現性幼稚至青春發育期無第二性徵發育,原發閉經。
3β-羥脫氫酶缺乏症
出生時即可出現失鹽和腎上腺皮質功能不全症狀,嚴重者因循環衰竭而死亡。男性為不同程度的外生殖器發育不全,如小陰莖,女性不同程度男性化、多毛、月經失調。
腎上腺危象
是一種威脅生命的突發狀況,典型臨床表現是脫水、低血壓、低鈉血症、高血鉀症、低血糖或精神症狀。若不治療或治療不及時,高血鉀症會導致猝死。
甲狀腺機能低下
先天性腎上腺增生症病人中觀察到的低血糖、低血壓、智力發育遲緩及心血管風險增加可能與甲狀腺激素的作用有關,但目前尚未查閱到明確的關於先天性腎上腺增生症與甲狀腺機能低下症之間的關係。
心肌損害
糖皮質類固醇不足可能在心肌損害中起一定作用,糖皮質類固醇的缺乏會導致心臟功能差、對兒茶酚胺的血管反應差,以及抗利尿激素分泌增加。
看醫
此病可用糖和鹽皮質激素替代治療,預防腎上腺危象發生。維持正常生長和骨骼成熟,力求達到理想的終身高。對家庭和患兒不良心理進行疏導。
- 對家族中有本病先證者的孕婦應做羊水細胞或者取絨毛膜進行產前基因診斷。
- 新生兒出生後第3天~5天,測定足跟血17-0HP 水平,篩檢先天性腎上腺增生症,已經確診者應積極治療並定時隨訪與監測。
大多病人優先考慮去兒科、兒童內分泌科、遺傳病專科就診。
- 因為什麼來就診的?
- 有無早篩?
- 目前都有什麼症狀?(如陰唇肥大、多毛、閉經等)
- 以前是否有其他內分泌科診治經歷?
- 既往有無其他的病史?
- 尿液17-羥類固醇 、17-酮類固醇和孕三醇測定:17-酮類固醇是反映腎上腺皮質分泌雄激素的重要指標,對本病的診斷價值優於17-羥類固醇。腎上腺增生症病人17-酮類固醇明顯升高。
- 血17-羥孕酮、腎素血管緊張素原、醛固酮、脫氫表雄酮、去氧皮質酮及睪固酮測定血17-羥孕酮、孕酮、脫氫表雄酮、睪固酮均可增高,其中血17-羥孕酮增高可為正常的幾十倍至幾百倍,是21-羥化酶缺乏症較可靠的診斷依據。
- 血皮質醇、促腎上腺皮質激素測定:典型失鹽型先天性腎上腺增生症病人的皮質醇水平低於正常,單純男性化可在正常範圍或稍低於正常。血促腎上腺皮質激素不同程度升高,部分患兒尤其不典型者可正常。
- 血電解質:失鹽型可有低血鈉、高血鉀症。
- 染色體檢查:外生殖器嚴重畸形時,可進行染色體分析,以鑑定性別。
- X線檢查:拍攝左手腕掌指骨正位片,判斷骨齡,病人骨齡常超過年齡。
- CT、磁振造影檢查:可發現雙側腎上腺增大。
- 基因診斷:採用直接聚合酶連鎖反應、寡核苷酸雜交、限制性內切酶片段長度多態性和基因序列分析可發現相關基因突變或缺失。
- 對於假兩性畸形、失鹽症群、高血壓或低血壓、低血鉀和鹼中毒表現的病人,均應考慮本病。
- 影像學檢查對腎上腺腫瘤有定位診斷價值:B 超發現雙側腎上腺回聲增強、增寬,則應進一步檢查。CT和MRI檢查表現為雙側腎上腺影普遍增大,邊緣略呈結節狀,但仍保持其大體形態,結構正常。
- 對胎兒或新生兒進行產前診斷可以CYP21缺陷症為目標,在懷孕後第2個3月期間進行。在第1個3月期,妊娠期9周或以後的絨毛膜切片檢查採樣,第2個3月期診斷以懷孕第 14週或以後行羊膜穿刺術。
- 新生兒篩檢主要用於典型CYP21D缺陷症的篩檢,測定出生24 h後足跟血液中 17-OHP,如升高可診斷70%的典型CYP21D缺陷症。
先天性肥厚性幽門狹窄症
表現為特徵性的噴射性嘔吐,鋇劑造影可發現狹窄的幽門,無皮膚色素沉澱,外生殖器正常。失鹽型易誤診為先天性肥厚性幽門狹窄或腸炎,故如遇新生兒反覆嘔吐、腹瀉,應注意家族史、生殖器外形等,必要時進行相關檢查。
真性性早熟
單純男性化型睪丸容積與實際年齡相稱,17-酮類固醇明顯升高,而真性性早熟睪丸明顯增大,17-酮類固醇增高,但不超過成人期水平。
男性化腎上腺腫瘤
男性化腎上腺腫瘤和單純男性化型均有男性化表現,尿17-酮類固醇均升高,需進行 Dexamethasone 抑制試驗,男性化腎上腺腫瘤不被抑制,而單純男性化型則顯示較小劑量 Dexamethasone 即可顯著抑制。
治療
先天性腎上腺增生症治療原則為糾正水、電解質不平衡,兒童首選氫化可的松或醋酸氫化可的松,有失鹽者需補充鹽皮質激素,藥物劑量應個體化,應激情況應加大腎上腺皮質激素藥物劑量。女性病人及失鹽型男女病人應終生治療,單純男性化型的男性病人在進入青春期和成年期後可酌情停藥,必要時切除增生的腎上腺。
腎上腺危象治療
糾正脫水,糾正低血鈉,補充生理鹽水,必要時補充3%高張鈉,9α-氟氫可的鬆口服、氫化可的松靜滴,一周後減量,3~4周后減至維持量。糾正嚴重高血鉀,如高血鉀難以糾正可予葡萄糖加胰島素靜滴。
應激狀態治療
因感染髮熱,體溫高於38.5℃時腹瀉伴有脫水、嚴重外傷、全麻手術,導致腎上腺衰竭的疾病等,增加皮質醇的劑量,輕度感染增加1~2倍,重度感染增加2~5倍,並同時控制感染。可以達到正常人在應激狀態時皮質醇增加的濃度,病情好轉後需迅速減回至維持治療用量。
- 皮質醇替代治療:採用氫化可的松或醋酸氫化可的松治療,補充腎上腺分泌皮質醇不足同時抑制垂體促腎上腺皮質激素的分泌,從而使雄激素的產生減少並阻止男性化的繼續發展,發揮正常生長潛能。糖皮質類固醇劑量應根據生長速率、骨成熟度、17-羥孕酮、睪固酮、促腎上腺皮質激素等指標調整。
- 鹽皮質激素治療:9α-氟氫可的松可協同糖皮質類固醇作用,使促腎上腺皮質激素分泌進一步減少。
- 腎上腺增生症時伴有腎上腺結節性腺瘤可考慮手術切除部分增生的腎上腺,後期腎上腺激素水平低可進行相應激素替代治療。
陰蒂退縮成形術
女孩陰蒂增大,需手術治療,最適宜手術年齡為2歲前,術前最好進行染色體核型的檢查,以確定遺傳性別。
陰道成形術
嚴重者需初期進行外生殖器矯形手術,應向家長說明在月經來潮之前還應確定陰道是否有先天畸形。
當治療過程中出現真性性早熟,骨齡明顯超速時,可聯合應用促性腺激素釋放激素治療,抑制中樞性性早熟,爭取骨骺晚閉合,儘量達到理想的終身高。
預後
先天性腎上腺增生症的患兒治療得當,兩性均可有正常的青春發育和生育功能。
本病大多需終身治療,單純男性化的男性病人進入青春期或成人,可適當停藥動態觀察。
治療得當一般不會影響自然壽命。
經過治療的病人應根據年齡和開始治療後的反應,開始需要每1~2月一次,劑量調整好後可3~6個月複查一次。
飲食
先天性腎上腺增生症患兒需食用富含維他命及高熱量的食物,失鹽型患兒每日的鈉攝入量不少於10 克~20 克,多喝水,維持身體水分。對於拒食的患兒需耐心餵養,必要時候可利用滴管或者鼻飼來滿足患兒的生理需要。
- 宜補充足量的魚肉蛋白維他命等,滿足身體基本需要。
- 對於嬰幼兒,注意及時添加輔食,可以添加少量多餐配方奶。
照護
先天性腎上腺增生症病人需要加強飲食管理,積極預防感染,預防腎上腺危象,積極進行心理干預與治療。
- 在激素應用過程中,應注意觀察激素的副作用,按時服用護胃藥、補鈣藥等。不要輕易停藥減藥,在發生應激性事件例如感染、手術等情況時,應遵醫囑適量調整激素用藥。
- 注意保暖,保持患兒全身皮膚乾燥清潔,同時加強對患兒的口腔護理,勤剪指甲,保持房內乾淨、通風。
- 觀察患兒精神情況,有無脫水、厭食、嘔吐、頭痛、嗜睡等不適,一旦發生腎上腺危象,立即就醫。
- 當患兒出現嘔吐症狀時,需抱住患兒頭部向前傾,用手扶住患兒的前額頭,讓嘔吐物吐出,以免嘔吐物進入呼吸道而引發窒息情況。
預防
先天性腎上腺增生症是常染色體隱性遺傳病,預防措施包括新生兒篩檢和產前診斷。
新生兒篩檢
主要對21-羥化酶缺乏症篩檢,目的是避免和預防延遲診斷治療造成的腎上腺皮質危象而導致的死亡。過多雄激素造成患兒日後身材矮小、心理生理髮育異常。
產前診斷
先天性腎上腺增生症是常染色體隱性遺傳病,每生育一胎就有1/4概率為先天性腎上腺增生症病人。因此,對家族中有本病先證者的孕婦應做羊水細胞或者取絨毛膜進行產前基因診斷。