
進行性脊肌萎縮症
概述
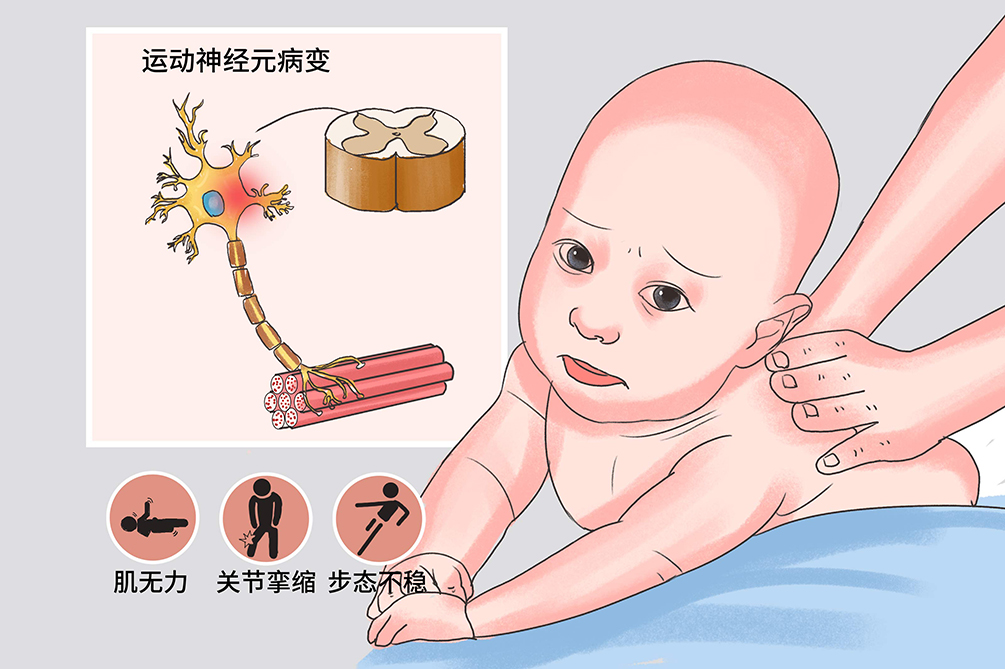
進行性脊肌萎縮症是由於運動神經元基因1突變導致SMN蛋白功能缺損所致的遺傳性神經肌肉病變,症狀主要表現為肌無力和肌萎縮。病理特點是脊髓前角細胞及腦幹運動神經核變性,是一組常染色體隱性遺傳性疾病。該病可分為四型,Ⅰ型病情進展迅速,無特殊治療。Ⅱ型和Ⅲ型、Ⅳ型應積極進行復健治療,以避免初期發生肌肉攣縮而殘廢。
- 就診科別:
- 神經內科
- 英文名稱:
- Progressive spinal muscular atrophy
- 疾病別稱:
- 進行性脊髓性肌萎縮症、脊髓性肌萎縮症
- 是否常見:
- 是
- 是否遺傳:
- 是
- 併發疾病:
- 關節畸形、肺部感染
- 治療周期:
- 終身治療
- 臨床症狀:
- 肌無力、腱反射減弱或消失、關節攣縮、步態不穩
- 好發人群:
- 染色體缺陷者、有脊肌萎縮症病家族病史者
- 鑑別診斷:
- 重症肌無力、重症佝僂病、強直性肌營養不良
- 常用檢查:
- 肌電圖、肌肉切片檢查、抗體檢測、基因診斷、血清CPK
目前公認分類為SMA-Ⅰ型、SMA-Ⅱ型、SMA-Ⅲ型以及SMA-Ⅳ型。大多數病人為Ⅰ型,其次為Ⅱ型,Ⅲ型發病率最低。
Ⅰ型
也稱為嬰兒型脊髓性肌萎縮症,本型在三型中最為嚴重,約1/3病例在宮內發病,胎動變弱。半數在出生時或出生後的最初幾個月即可發病,且幾乎均在5個月內發病。肌肉病理特徵是存在著大組分布的圓形萎縮肌纖維,常累及整個肌束;亦見肥大纖維散在分布於萎縮纖維之中,兩型纖維均可受累。
Ⅱ型
也稱為少年型SMA、中間型SMA或慢性SMA,發病較Ⅰ型稍遲,多於1歲內起病。一些年齡稍大,進入相對穩定期的患兒,可出現繼發性肌性損害改變,包括中央核增多以及肌纖維撕裂等現象。
Ⅲ型
是SMA中表現最輕的一類,本病在兒童末期或青春期出現症狀,表現為神經元性近端肌萎縮,容易和肢帶型肌營養不良相混淆。本型肌纖維肥大改變十分突出,直徑可達100~150μm,常合併繼發性肌原損害。包括纖維撕裂、中央核改變、NADH染色見蛾噬樣及指紋狀纖維、少量壞死和再生纖維、巨噬細胞浸潤以及間質脂肪結締組織增生等。
Ⅳ型
發病時間在成年初期,預後較好。症狀與Ⅲ型相似。
病因
進行性脊肌萎縮症是一種常染色體隱性遺傳疾病,主要病因為SMN1基因突變導致脊髓前角運動神經元和腦幹運動神經核變性,無傳染性。有該病家族史的即攜帶該基因的兒童容易發病,可在母體妊娠期通過基因診斷來鑑定胎兒是否健康。
本病為遺傳性疾病,為SMN1基因突變所致。基因的突變以及轉化引起脊髓前角運動神經元和腦幹運動神經核變性,最終導致進行性脊肌萎縮症的發生。
進行性脊肌萎縮症是一種常見的常染色體隱形遺傳疾病,發病率約為1/10000~1/6000,攜帶率為1/50~1/40。
兒童
由於本病為基因類疾病,發病較早,尤其是SMA-第一型,有三分之一病人在子宮內就會發病。
有家族史病人
本病為遺傳性疾病,家族中有該病基因攜帶者,生下的後代中更容易出現本病。
症狀
進行性脊肌萎縮症的病人典型症狀為肌無力、以及肌肉無力帶來的運動障礙、吞咽困難、呼吸系統感染、關節畸形、發育遲滯等現象。除此之外,一些病人還有因為肌肉無力影響正常的呼吸、行走,導致呼吸感染、步態異常、關節畸形、脊柱側彎等情況。嚴重者會喪失正常的自理能力,社會交往能力和運動功能也明顯喪失。
- 嚴重肌無力、吸吮能力弱、吞咽困難、呼吸道感染、呼吸衰竭。
- 發育遲滯、運動功能障礙、多發性微小陣攣。
- 近端肢體無力,骨盆比肩部嚴重,上下樓梯困難。
- 肌力下降、關節畸形,嚴重者會發生脊柱側彎,發病時間在成年期。
- 多數病例表現以近端為主的嚴重肌無力,下肢重於上肢,需要在幫助下行走,不能獨自完成行走。
- 由於呼吸功能不全和容易發生誤吸,任何輕微的上呼吸道感染可迅速演變為重症肺炎,危及生命。病人的社會交往能力和運動功能喪失明顯。
呼吸道、肺部感染
由於肋間肌和膈肌的肌無力,引起通氣不足以及咳嗽微弱,長期臥床可造成墜積,誤吸也可造成肺炎。即使在沒有呼吸道或者是肺部感染的情況下,病人也需保持良好的肺部通氣狀態,預防發生進行性肺不張,導致病人死亡。可輔助咳嗽、胸部叩擊治療及間歇正壓通氣預防。
脊柱側彎
脊柱側彎是進行性脊肌萎縮症最為嚴重的骨骼畸形,不能行走的病人會更早地發展為脊柱畸形,大部分的畸形位於胸腰部。且脊柱矯正法常不能預防或延緩脊柱側彎,需要嚴密監測脊柱側彎情況,防止因側彎嚴重導致的呼吸衰竭。
營養不良和生長障礙
病人常由於吮吸乏力、氣道不暢或容易疲勞導致營養不良和生長障礙。患兒由於負氮平衡會導致肌無力和疲勞加重,尤其是嬰兒。這種現象的機制目前尚未闡明。部分病人由於營養攝入不足導致有機酸尿症,故需要有專業的營養師對其進行餵養指導。
便秘
由於行動障礙,有些病人會發生便秘,但通過增加液體和纖維攝入,可減輕便秘。
看醫
進行性脊肌萎縮症出生後最早表現出的症狀在肌肉無力以及關節活動範圍的異常上,大多數病人發病年齡較小,會去兒科就診。且該病屬於遺傳類疾病,如果家中有該病病史的病人,那進行妊娠時應該進行詳細的基因診斷,如果胎兒存在SMN1基因,應慎重考慮。
無論何種情況,有關節活動範圍或者形態異常和肌肉無力的患兒都應進行就醫。該病為進行性疾病,一開始症狀表現輕微,後逐漸加重,應在疾病剛發現時立即就醫,爭取及早控制。
- 由於發病年齡關係,大多數病人在兒科就診。
- 若病人發病時已經成年,可以去神經內科就診,如果需要進行相應的鍛煉可以去復健科就診。
- 有沒有出現關節攣縮?
- 什麼時候發現病情的?
- 目前肌力情況怎麼樣
- 有沒有出現吞咽障礙?
- 有沒有出現過肺炎?
- 有沒有呼吸道感染病史?
- 身體骨骼有沒有出現畸形?
- 除了肌肉無力有沒有其他症狀?
基因診斷
可通過血DNA分析檢測SMN基因突變,從而診斷疾病。一旦發現SMN基因突變,則不需要再作其他檢查,即可確診為進行性脊肌萎縮症。
血清肌酸磷化酶
Ⅰ型正常,Ⅱ型偶見增高。Ⅲ型常增高,甚至可達正常值10倍以上,同工酶變化以MM為主,隨著肌損害的發展而增加,至末期肌肉萎縮時,血清肌酸磷化酶才開始下降,這與肌營養不良不同,後者於嬰幼兒期即達到高峰,以後漸降。
電生理檢查
電生理檢查可反映SMA的嚴重程度,電生理檢查NCV示運動傳導速度可減慢,在Ⅰ型減慢,而其他類型正常;感覺傳導速度正常。纖顫電位及正銳波在各型SMA均可出現,但SMA-Ⅰ型更明顯。隨意運動時,各型SMA均見干擾相減少,尤其是Ⅰ型SMA僅呈單相。在較末期Ⅲ型SMA可見類似於肌源性損害的低波幅多相電位。
肌肉活體組織檢查
肌肉活體組織檢查對確診SMA具有重要意義,其病理表現特徵是具有失神經和神經再支配現象。
基因診斷中發現SMN基因突變,不需要再做其他任何檢查,即可確診為進行性脊肌萎縮症。
重症肌無力
骨骼肌的肌無力的波動性和疲勞性。肌無力表現為活動後加重、休息後減輕的隨意肌無力,多為晨輕暮重。進行性脊肌萎縮不存在波動性。
重症佝僂病
除肌肉無力、生長遲滯外,可有神經系統發育遲緩,語言發育落後,免疫功能下降,反覆感染等症狀,且經治療可改善,進行性脊肌萎縮症無有效治療方法,治療後效果不佳。
強直性肌營養不良
主要症狀為進行性肌肉無力萎縮和肌強直現象。同時伴有多系統受累症狀,如心律失常、早發白內障、初期額禿、生育能力較低、輕度認知及睡眠障礙等。常20~40歲出現臨床表現。進行性脊肌萎縮症多在胎兒期、嬰幼兒期出現。
治療
進行性脊肌萎縮症無特殊治療方法,不能徹底治癒,只能通過復健治療控制疾病症狀。本病Ⅰ型病情進展迅速,無特殊治療。Ⅱ型和Ⅲ型應積極進行復健治療,以避免初期發生肌肉攣縮而殘廢。
- 進行性脊肌萎縮症可根據病人情況,進行肌力鍛煉,增強病人肌力。關節活動度鍛煉,保持關節關節活動度範圍。
- 對於關節沾黏攣縮病人可用Ⅳ級鬆動手法,松解沾黏組織。面肌無力影響吞咽病人,可行吞咽功能訓練,鍛煉病人口臉部肌肉以及唇舌部肌肉,改善病人吞咽能力,防止病人誤吸。
- 對於關節畸形病人,可應用矯形器,畸形輕度病人鍛煉相應肌群,矯矯正牙齒形。
- 對於呼吸肌無力病人可用膈肌起搏器以及呼吸功能訓練,如縮唇呼吸等,鍛煉呼吸功能和心肺功能,減少肺炎發生。
進行性脊肌萎縮症無藥物治療。
進行性脊肌萎縮症無手術治療。
預後
進行性脊肌萎縮症預後不佳,目前無法治癒,只能通過各種手段延長生存時間提高生存質量。到目前為止,沒有較好的藥物與手術可用於該病的治療。
進行性脊肌萎縮症為終身疾病,目前尚不能治癒。
本病Ⅰ型患兒生存期約1年左右,Ⅱ型病程進程較緩慢,可以存活至少年期。Ⅲ型、Ⅳ型可如果接受較好的臨床干預可生存至25歲,部分病人甚至可以達到正常壽命。
進行性脊肌萎縮症可根據發病時間制定複診,剛發病時需3個月複診一次,待到病情逐漸穩定,症狀不會加重後,可6個月複查一次。
飲食
進行性脊肌萎縮症病人應注意膳食多樣化、增強膳食營養,必要時可使用鼻飼管進食。多吃富含維他命B的食物,進行合理的飲食規劃。對待面肌無力病人,注意飲食時防止誤吸。
照護
進行性脊肌萎縮症病人的護理以預防關節畸形和各種感染為主,加強日常生活管理。注重病人營養健康與心理健康,給予病人充分關愛,對年幼病人加強監控,防止因呼吸道感染或骨骼畸形出現的意外。
- 保持生活環境潔淨、空氣清新,使病人心情舒暢。
- 大多病人發病年齡較小,需要注重病人心理健康,對病人進行心理治療。家人要給予病人支持,使病人感到家人的關愛,增強生存信心。
該病為進行性疾病,會逐漸加重,嬰幼兒需要加強呼吸監控,防止故意感染,或者由於骨骼畸形導致的窒息。青少年需加強骨骼畸形的監控,3個月去檢查一次,必要時可使用矯形器。
預防
目前進行性脊肌萎縮症沒有較好的預防方法,只能通過產前篩檢來進行風險評估,之後決定是否進行胎兒妊娠。
家庭成員應進行攜帶者篩檢並在孕前進行遺傳諮詢和風險評估,高危妊娠者可進行胎兒的產前診斷。進行性脊肌萎縮症的產前診斷應採用基因診斷技術。當確認胎兒攜帶有與先證者SMN1基因相同突變時,提示是患胎,應在知情的情況下,由其雙親決定是否繼續妊娠。
進行性脊肌萎縮症為遺傳性疾病,無較好的預防措施,可在妊娠時進行基因檢測,確定胎兒是否出現SMN1相同基因,如出現胎兒存在該病,如不存在則為健康胎兒。