
遺傳性小腦萎縮症
概述
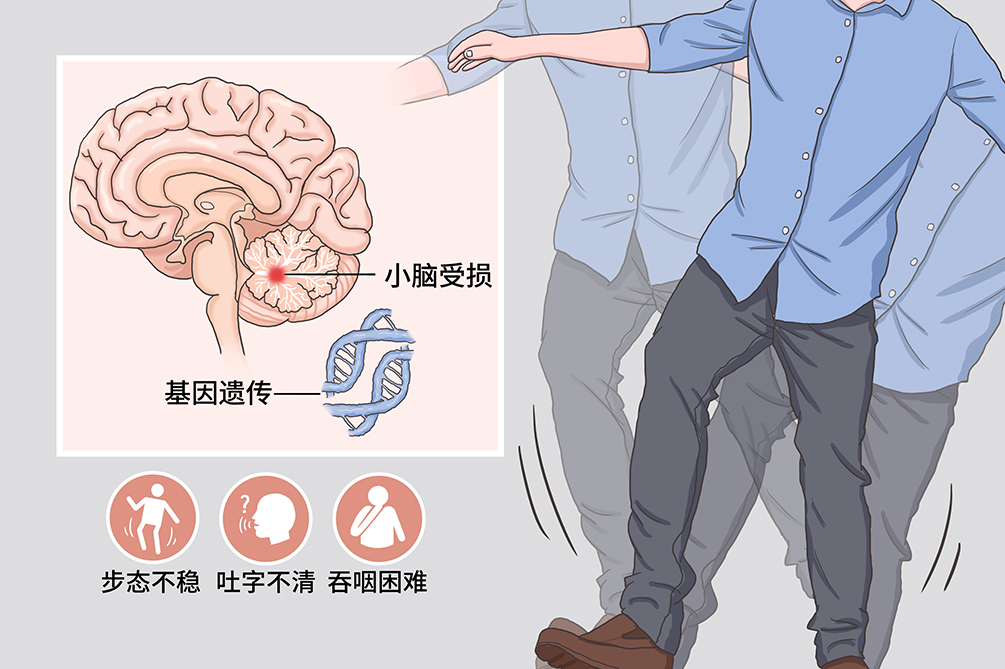
遺傳性小腦萎縮症是常染色體異常引起的共濟運動障礙為突出表現的中樞神經系統變性疾病。本病進展緩慢、有家族遺傳史。本病較罕見,臨床表現為平衡障礙、進行性肢體協調運動障礙、步態不穩、構音障礙、眼球運動障礙等,並可伴有複雜的神經系統損害,如錐體系、錐體外、視覺、聽覺、脊髓、周圍神經損害,亦可伴大腦皮質功能損害,如認知功能障礙和(或)精神行為異常等。該病因尚不明確,大都有遺傳史、家族史,具有家族系譜。目前治療主要採用對症和支持治療,本病的死亡率和致殘率較高。
- 就診科別:
- 神經內科
- 英文名稱:
- hereditary ataxia(HA)
- 是否常見:
- 否
- 是否遺傳:
- 是
- 併發疾病:
- 心臟病變、內分泌代謝異常、骨骼畸形、皮膚病變
- 治療周期:
- 長期持續性治療
- 臨床症狀:
- 痙攣、顫抖、認知功能障礙
- 好發人群:
- 20~40歲人群
- 常用藥物:
- 5-羥色胺1A受體激動劑、 Carbamazepine 、 Levodopa 、維他命E、輔酶Q10
- 常用檢查:
- CT、磁振造影、神經電生理檢查、實驗室檢查
根據小腦萎縮症類型的不同分為三類
脊髓型
- Friedreich型小腦萎縮症:常染色體隱性遺傳,青少年起病,初始行走不穩,漸出現後索損害的症狀,Romberg征(+),睜眼可以改善。繼之脊髓小腦束受累,出現步基寬、蹣跚步態、定向性顫抖和小腦性構音障礙。肢體肌肉張力降低,腱反射減低或消失,下肢沉重。部分病人可伴有弓形足、脊柱側彎及其他畸形,個別病人可有心臟異常。
- 遺傳性痙攣性截癱:常染色體顯性或隱性遺傳或性連遺傳。兒童期起病,男性多見,主要為錐體束受損,多為下肢呈緩慢加重的痙攣性癱、剪刀狀步態。無感覺障礙,上肢很少受累,可伴有原發性視神經萎縮或視網膜色素變性。
小腦型
- Marie型小腦萎縮症:常染色顯性遺傳,成年起病,自下肢開始出現小腦型小腦萎縮症而無感覺障礙,言語常頓挫或暴發,可有錐體束征及欣快,智力減退。
- 橄欖小腦橋腦萎縮:常染色體顯性遺傳,中年後起病,除小腦型小腦萎縮症和構音障礙外有初期尿失禁,部分病人有智能減退和錐體外症狀,如帕金森氏症等,但無眼球顫抖。
脊髓小腦型
主要有小腦萎縮症-微血管擴張症。常染色體隱性遺傳,嬰兒期發病,小腦型小腦萎縮症,構音障礙,皮膚、顏面微血管擴張,多數伴有舞蹈樣手足徐動,隨年齡增長而明顯。青春期後出現深感覺消失等脊髓後索症狀,和病理反射陽性。可因免疫缺陷而反覆發生呼吸道感染,末期有肺部廣泛纖維化、肺功能不全等。
按遺傳類型分類可分為以下2種
常染色體顯性遺傳
- 橄欖-腦橋-小腦萎縮Ⅰ、Ⅲ、Ⅳ、Ⅴ型。
- Machado-Joseph’s disease(MJD)。
- 小腦實質變性Ⅰ型和Ⅳ型。
- 脊髓腦橋變性。
- 遺傳性痙攣性截癱。
- 發作性小腦萎縮症。
- 後索性小腦萎縮症。
常染色體隱性遺傳
- 橄欖-腦橋-小腦萎縮Ⅱ、Ⅵ型。
- 小腦實質性變性Ⅱ、Ⅲ、Ⅳ型。
- 肌陣攣性小腦性小腦萎縮症,或稱Ramsay-Hunt症候群。
- 小腦萎縮症性微血管擴張症。
- Friedreich型小腦萎縮症。
病因
目前該病病因和發病機制尚未闡明,認為可能主要與以下遺傳因素有關,如酶缺乏、缺陷,生化缺陷、線粒體功能缺陷、DNA修復缺陷。
酶缺乏、缺陷
多見於丙酮酸脫氫酶複合體、氨基己糖苷酯酶A、鳥氨酸氨甲醯基轉移酶等缺乏。
生化缺陷
如小腦萎縮症伴選擇性維他命E缺乏症中的維他命E缺乏,如β-脂蛋白缺乏症中的β-脂蛋白缺乏。血棘紅血球增多和血VE低,如Hartnup,病中的胺基酸尿症。
三核甘酸動態突變
突變形式可表現為突變無義、缺失突變。動態突變是一種新型的突變形式,其是由於DNA中的基重複單位拷貝數不穩定擴增而導致。正常情況下,重複單位拷貝數有一定限制,而在動態突變下,則大大增加。
線粒體功能缺陷
線粒體是給細胞提供能量的細胞區器。線粒體病是一組由MTDNA或核DNA(NDNA)缺陷導致MT結構和功能障礙,ATP合成不足所致的多系統疾病。線粒體病是由核或基因缺陷引起。
DNA修復缺陷
與DNA修復有關的小腦萎縮症有AT、著色性干皮病(xP)、Cockayne症候群。
據統計,遺傳性小腦萎縮症本病約占整個神經遺傳病的10%。大多數在20~40歲發病,嬰幼兒和老人發病較少。其遺傳方式以常染色體顯性遺傳方式為最多。
該病好發於20~40歲人群。
症狀
該病主要表現為共濟運動障礙,伴有複雜的神經系統損害。神經系統表現包括平衡障礙、進行性肢體協調運動障礙、步態不穩、構音障礙、眼球運動障礙等,並可伴有複雜的神經系統損害,如錐體系、錐體外、視覺、聽覺、脊髓、周圍神經損害,亦可伴大腦皮質功能損害,如認知功能障礙和(或)精神行為異常等。還可以出現神經系統以外的病變,如心臟病變、內分泌代謝異常、骨骼畸形、皮膚病變等。
步態不穩
是最常見的首發症狀,出現醉酒樣或剪刀步伐。
吐字不清
構音障礙為遺傳性小腦萎縮症的特徵之一,病人主要表現為發音生硬(爆發性言語)、緩慢,單調而含糊,構音不清、音量強弱不等,或時斷時續,呈吟詩樣語言。病情進展至末期時,幾乎所有病人均出現運動失調性構音障礙。
吞咽困難和飲水嗆咳
是由於腦幹神經核團受損所致,隨著病情的進展,臨床表現逐漸明顯且多見。
顫抖
主要表現為運動性顫抖、姿勢性顫抖或意向性顫抖,若伴有錐體外損害,也可出現靜止性顫抖。
痙攣狀態
由錐體束受損所致,表現為軀幹及肢體肌肉張力增高、腱反射活躍或亢進、踝陣攣、Babinski征陽性等,行走時呈明顯的痙攣性步態。
錐體外症狀
部分病人由於基底節受損,故可伴發帕金森氏症樣表現,或出現面、舌肌搐顫,肌陣攣、手足徐動症、扭轉痙攣、舞蹈樣動作等錐體外表現。
認知功能及精神障礙
表現為注意力、記憶力受損,任務執行功能下降,其中憂鬱、睡眠障礙、精神行為異常、偏執傾向是臨床常見的精神障礙。
- 視神經病變:原發性視神經萎縮、視網膜色素變性等症狀可見於常染色體顯性遺傳性小腦萎縮症Ⅱ型、Friedreich小腦萎縮症、小腦萎縮症⁃微血管擴張症、植烷酸貯積病等亞型,病人多伴有視力、視野及瞳孔改變。
- 骨骼畸形:為常見體徵,主要表現為脊柱側彎或後側凸,少數病人還可發生爪形手或隱性脊柱裂等畸形,尤其是Friedreich小腦萎縮症病人,以弓形足及脊柱彎曲最常見。
- 皮膚病變:多見於眼球結膜、面頸部皮膚微血管擴張、皮膚魚鱗症、牛奶咖啡色素斑等表現,常見於小腦萎縮症-微血管擴張症或Refsum症候群病人。
- 神經系統外的其它症狀還包括心肌肥厚、糖、脂肪酸、磷脂、維他命代謝異常。
看醫
大部分病人因為步態不穩、吐字不清、吞咽困難和飲水嗆咳、顫抖、軀幹及肢體肌肉張力增高、腱反射活躍或亢進,面、舌肌搐顫,肌陣攣、手足徐動症、扭轉痙攣、舞蹈樣動作就診,通過實驗室檢查、影像學檢查、神經系統檢查等進行診斷。
出現步態不穩、吐字不清、吞咽困難和飲水嗆咳、書寫障礙、站立不穩,注意力、記憶力受損,憂鬱、睡眠障礙、精神行為異常、偏執傾向等,尤其是有遺傳性小腦萎縮症家族史病人,需要及時就醫。
該病病人應優先考慮至神經內科就診。
- 症狀是什麼時候出現的?
- 目前都有什麼症狀?(如步態不穩、吐字不清、吞咽困難和飲水嗆咳、書寫障礙、站立不穩等)
- 有無遺傳性小腦萎縮症家族史?
- 是否有既往病史?
- 是否去其他醫院就診過?
神經系統檢查
進行小腦功能、肌肉力量、感覺、反射等方面的檢查。
影像學檢查
CT、磁振造影可顯示是否出現小腦或腦幹萎縮,病人一般出現頸髓萎縮。
實驗室檢查
包括血糖、脂肪酸、磷脂、維他命代謝等檢查,但以下遺傳性小腦萎縮症病人伴有特異性的生化指標異常。
- 小腦萎縮症伴肌陣攣或肌陣攣癲,包括線粒體腦肌病、蠟樣脂質褐質沉積病(ceroid lipofuscinosis)、唾液酸沉積症(sialidosis)等。
- 威爾森氏症(HLD),有些威爾森氏症病人小腦體徵十分顯著,血清銅藍蛋白檢測有助於診斷。
- β脂蛋白缺乏症,與維他命E吸收障礙有關,但隨著年齡的增長其症狀可逐漸減弱。光學顯微鏡下常可發現棘紅血球,且血清中不能檢測到β脂蛋白。
- 腦腱黃瘤病,以年輕人好發,主要表現為痙攣-小腦萎縮症症候群、動脈粥狀硬化、白內障。腱黃瘤的存在和血清高膽甾烷醇水平有助於診斷顱內可能存在的黃瘤。
神經電生理檢查
可檢測出病人是否出現體感誘發電位、聽覺誘發電位、視覺誘發電位、眼震電圖、神經肌電圖等異常。
有遺傳性小腦萎縮症的家族史
醫生會詢問病人是否有該病家族史,幫助進行疾病診斷。
神經系統表現
包括步態不穩、吐字不清、吞咽困難和飲水嗆咳、書寫障礙、站立不穩等,亦可伴大腦皮質功能損害,如認知功能障礙和(或)精神行為異常等。還可以出現神經系統以外的病變,如心臟病變、內分泌代謝異常、骨骼畸形、皮膚病變等。
CT、磁振造影提示小腦萎縮
有時可見腦幹萎縮,腦幹誘發電位可出現異常,肌電圖顯示周圍神經損害。
實驗室檢查
血糖、脂肪酸、磷脂、維他命代謝,部分病人可出現異常。
神經電生理檢查
出現眼震電圖、神經肌電圖等異常,幫助該病進行診斷。
遺傳性痙攣性截癱
遺傳性痙攣性截癱病人可有小腦萎縮症障礙等神經系統表現,可通過基因檢測與遺傳性小腦萎縮症進行鑑別。
中毒性小腦萎縮症
病人有小腦萎縮症障礙,多由於酒精、藥物、重金屬所致,且無家族遺傳史。而遺傳性小腦萎縮症有家族史,可以此進行鑑別。
治療
目前尚無能夠完全阻止遺傳性小腦萎縮症病情進展的治療,遺傳性小腦萎縮症的臨床治療仍以對症治療和支持治療為主,主要目標是減輕症狀、緩解病情進展、維持日常生活自理能力。
5-羥色胺1A受體激動藥改善小腦萎縮症症狀
如 Buspirone 可部分改善輕度小腦小腦萎縮症症狀, Tandospirone 治療脊髓-小腦性小腦萎縮症3型部分有效。
Levodopa
可通過血⁃腦脊髓液屏障進入中樞神經系統,經多巴脫羧酶作用轉化為多巴胺從而改善肌強直、運動減少等症狀。 Trihexyphenidyl 對中樞神經系統膽鹼受體有阻斷作用,可改善肌強直、運動減少等症狀。
抗癲癇藥物
抗癲癇藥物 Carbamazepine 可較好控制病人的癲癇發作症狀。目前,對於病人所伴隨的構音障礙症狀尚無有效的對症治療藥物,可通過言語矯正訓練進行改善。
擴張血管和改善循環藥物
可擴張外周血管,改善血液循環,如煙酸、維他命E、煙酸酯、環扁桃酯、己酮可可鹼等。
神經元活化藥物
輔酶Q10可促進神經元代謝和呼吸功能,促進氧化磷酸化,具有抗氧化、保護生物膜結構完整性的功效。
維他命類藥物
對維持神經元正常代謝過程和改善功能有一定作用。可服用B群維他命、維他命E等。
本病無手術治療。
主要採取認知治療,以改變病人非理性信念,改善認知曲解及負性思維,喚起病人的正性情感,使其發揮自身能動性。
認知功能障礙
目前尚無有效的藥物治療,對病人初期的心理治療策略包括認知行為干預治療,有助於症狀出現後的積極應對。
神經復健
步態不穩的病人可以接受平衡功能訓練,幫助病人恢復正常生活。
預後
該疾病目前不可治癒,發現疾病時應及時延緩病情進展,緩解臨床症狀,維持日常生活自理能力。部分病人需要終身進行復健治療。
該病目前不可以治癒。
該病會影響自然壽命。
任何病人均需進行長期遵醫囑隨訪,部分病人需要終生復健治療。
飲食
該疾病並無特殊飲食禁忌,正常飲食即可。
照護
遺傳性小腦萎縮症病人需注意休息,少熬夜,保持良好的生活習慣,注意改善生活環境,家屬應加強與病人交流,對病人進行相應的心理疏導,注意保持衛生。
- 注意保持衛生,改善生活環境。
- 加強與病人交流與心理指導。
日常應注意監測病人步態不穩、吐字不清、認知功能障礙等症狀是否有加重,如有加重需及時到醫院就醫。
患有該病的病人在日常生活中需特別注意,需要別人照顧,注意摔傷及跌倒。
預防
遺傳性小腦萎縮症預防的重點是進行遺傳諮詢,產前診斷或胚胎植入前診斷是目前控制遺傳性小腦萎縮症發病的最佳手段。
孕前需要進行遺傳諮詢。
- 避免近親結婚,攜帶者需要進行基因檢測。
- 產前診斷和胚胎植入前診斷是目前控制該疾病的最佳手段。